

Applied mathematics and data science to tackle infectious diseases
See descriptions of our research on the following topics:
We developed compartmental models that incorporate both reported and unreported infectious individuals in the COVID-19 outbreak. These models were used to analyse strategies to suppress the virus in Germany, the Hubei province of China, Italy, Spain and the UK. The research provided evidence to confirm that: (i) more than 50% of infectious individuals were not tested for infection at the early stages of the epidemic, (ii) reducing the underlying transmission of untested cases was crucial to suppress the virus, and (iii) establishing herd immunity was not feasible during the early months of the epidemic.
- Learn more: Paper
We used data on the incidence of COVID-19 from across the world, together with student and staff numbers at UK HEIs in a stochastic mathematical model to predict that 81% of the UK HEIs had more than a 50% chance of having at least one COVID-19 case arriving on campus at the beginning of the 2020-2021 academic year. Predictions for the number of cases expected at each campus were also provided. Based on these estimates, it was suggested that universities had to plan for COVID-19 cases to arrive on campus and facilitate mitigations to reduce the spread of disease particularly during the first two weeks of term.
- Learn more: Paper.
Policymakers require consistent and accessible tools to monitor the progress of an epidemic and the impact of control measures in real time. One such measure is the Estimated Dissemination Ratio (EDR), a straightforward, easily replicable, and robust measure. It is comparable to the commonly used reproduction number, but simpler to calculate and explain. In collaboration with public health teams in Scotland, we used the EDR to monitor the progression of the COVID-19 outbreak in the UK. The EDR can demonstrate changes in transmission rate before they may be clear from the epidemic curve. Thus, EDR can provide an early warning that an epidemic is resuming growth, allowing earlier intervention. The EDR is comparable to the commonly used reproduction number, but easier to estimate.
- Learn more: Paper.
- Press coverage of our work on COVID-19: UoA News, STV, NorthSound 1 Radio, The Times, Daily Mail, The Evening Express, The Hippocratic Post, Express and Star, Yahoo UK, Medical X press, Gibraltar Chronicle, ...
Attributing or assigning individuals to a source population is important within many disciplines including ecology, anthropology, infectious diseases and forensics. A common strategy to attribute individuals to populations consists in comparing the genotype of the individual with the genetic profiles of defined source populations. We use data in which individuals of known source are characterised by genetic markers (genes, SNPs, etc) to train classifiers that can be used for source attribution of individuals whose source is unknown.
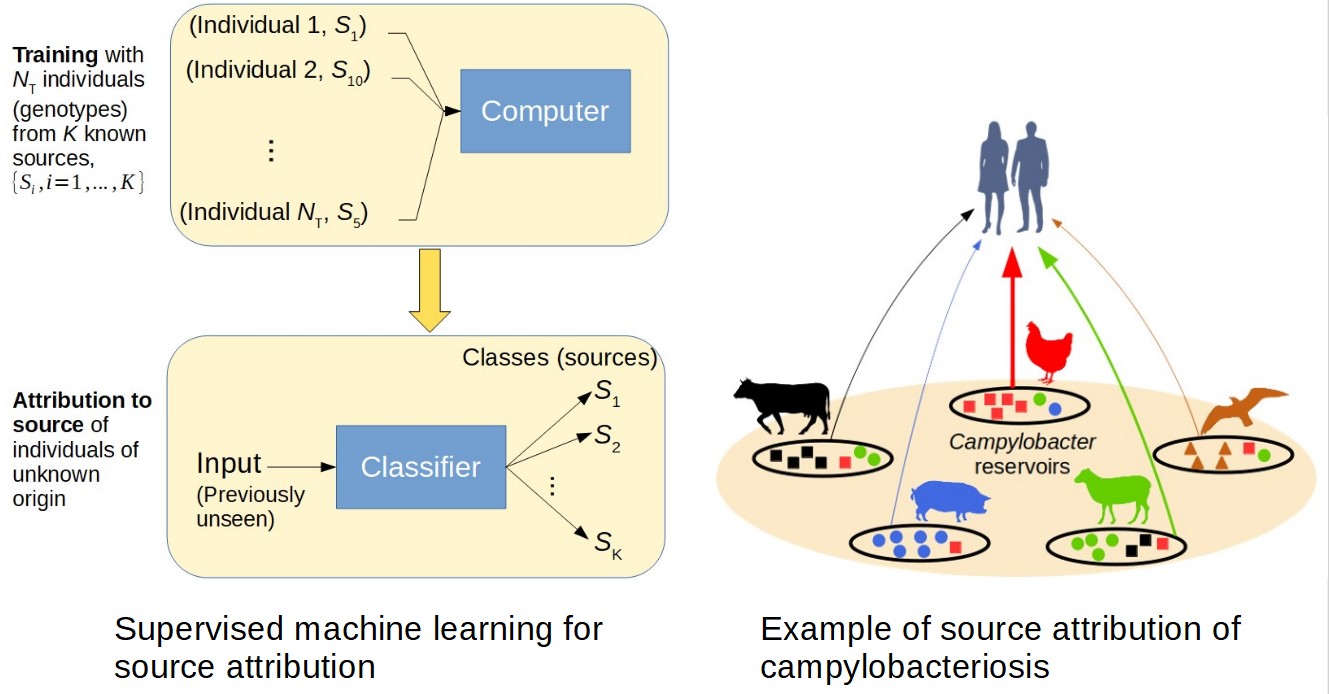
We developed a new machine learning method to attribute individuals to sources: The Minimal Multilocus Distance (MMD) method. This method is computationally fast and can operate on many thousands of genomic markers. In addition, the MMD method is generic, easy to implement for Whole genome sequence (WGS) data proteomic data and has wide application.
- Download our open source software as an executable version for Windows or as an R package.
- Learn more: Paper .
- Press coverage: UoA News, Food Safety News, Evening Express, The Hippocratic Post, Phys.org, Australian Institute of Food Safety, Laboratory Equipment, digit.fyi, ...
Within the context of infectious diseases, we have mostly focused on source attribution of Campylobacter and Listeria infections.
Campylobacter is the most common cause of bacterial food poisoning in the world. Using source attribution models, we identified shop-bought chicken meat as the main source of human campylobacteriosis (see, for example, our papers in Clinical Infectious Diseases and Scientific Reports).
Listeria causes around 2,500 infections and 250 deaths per year in the EU. In a joint project with Statens Serum Institut (SSI), French Agency for Food, Environmental and Occupational Health & Safety (ANSES) and Public Health England (PHE), we used source attribution to identify that the main source of listeriosis was food of bovine origin but there were also contributions from other food animals, including fish (see more information in this report for the European Food Safety Authority (EFSA)).
We extended the explosive percolation paradigm to develop “explosive immunisation”: a new method to inform targeted immunisation that enables the burden of epidemics to be minimised by vaccinating as few people as possible. The method assigns a score to each individual to quantify their contribution to the chance of a large epidemic if they are not immunised. Explosive immunisation targets individuals with a high score, who were called ‘superblockers’. These are typically well-connected individuals who move between different communities and whose immunisation abruptly decreases the possibility of an epidemic.
- Learn more: Paper
- Press coverage: UoA News, The Times, The National, Phys.org, R&D World, vesti.ru, ...
- Download open source software from GitHub.
We developed compartmental epidemic models to account for local synergistic effects observed in several phenomena including fungal invasion or the spread of social content. With these models, we found that synergistic effects associated with acquaintances of pairs of individuals can explain why both interesting and dull social content can become popular overnight, in a more explosive manner than epidemics.
- Learn more: Paper
- Press coverage: UoA News, The Times, The Scotsman, Daily Mail, The Sun, The Herald Scotland, The Independent Ireland, Phys.org, 20 Minutos, cubadebate, ...
- Related contribution to The Scotsman 200 Voices.
We have applied Quantitative risk assessment (QRA) to food safety challenges since 2008. QRA is a mathematical methodology employing Monte Carlo simulation, that estimates the risk of disease following consumption of a contaminated food. Here, we describe some of our work on the risk of foodborne disease associated with the pathogens E. coli O157 and Anisakis.
Escherchia coli (STEC) O157:H7 is a zoonotic pathogen that causes numerous food and waterborne disease outbreaks. We developed dose-response models, a key component of QRA, for E. coli O157 under Bayesian frameworks utilising human outbreak data (see, for instance, our paper in Epidemiology & Infection). We discovered that there is considerable heterogeneity across the virulence of E. coli O157 strains but that ingestion of 100-200 bacteria is sufficient for 50% of infected people to fall ill, posing a considerable health risk to consumers. We used these models to assess the risk of E. coli O157 infection from beef-burgers, private water supplies and direct contact with infected animals.
Anisakiasis is an emerging zoonosis caused by the fish parasitic nematode Anisakis. Using data on fishery landings, fish infection rates and consumption habits of the Spanish population from questionnaires, we developed a quantitative risk assessment (QRA) model for the anchovy value chain. Spaniards were estimated to consume on average 0.66 Anisakis per untreated (non-frozen) raw or marinated anchovy meal. The number of annual anisakiasis cases requiring medical attention was predicted between 7,700 and 8,320. Monte Carlo simulations estimated post-mortem migration of Anisakis from viscera to flesh increases the disease burden by >1000% whilst an education campaign to freeze anchovy before consumption may reduce cases by 80%. The QRA suggests that previously reported figures of 500 anisakiasis per year in Europe is a considerable underestimate.
- Learn more: Paper
- Press coverage: El País, El Mundo, El Confidencial, Scientific American, MedTempus, ...
The zoonotic pathogen E. coli O157:H7 is globally distributed and we explained its origin and the temporal sequence of its geographical spread. This was done by combining Monte Carlo based Bayesian methods with whole genome sequencing (WGS) and geographical information. We found that this virulent pathogen originated in the Netherlands around 1890 and spread around the world by >30 transmission events likely facilitated by farm cattle movements.
- Learn more: Paper
- Press coverage: New Zealand Herald, Australasian Science, The Conversation, ...